희귀질환은 다양하고 복잡한 특성을 가지며 전 세계적으로 수천 가지의 희귀질환이 알려져 있습니다. 여기서는 가장 대표적인 희귀질환 몇 가지를 소개하겠습니다.
1. 프레더릭 신경병증 (Friedreich's Ataxia)
프레더릭 신경병증(Friedreich's Ataxia, FA)은 유전적으로 발생하는 희귀 신경근육 질환입니다. 이 질병은 주로 신경계와 심장에 영향을 미치며, 조기 발병과 점진적 악화가 특징입니다.
원인
프레더릭 신경병증은 FXN 유전자의 돌연변이로 인해 발생합니다. 이 유전자는 주로 미토콘드리아에서 발견되는 필수 단백질인 프라타민을 인코딩하는데, 돌연변이로 인해 프라타민의 생산이 부족해지면 세포의 에너지 생산에 문제가 발생합니다. 이는 신경 세포와 심장 세포에 특히 치명적입니다.
증상
프레더릭 신경병증의 증상은 다음과 같습니다:
- 운동 조정 능력 저하 (아타시아): 이는 가장 흔한 초기 증상으로, 보행이 불안정해지고, 손의 정교한 움직임이 어려워집니다.
- 근육 약화: 다리, 팔, 그리고 몸통의 근육이 약해집니다.
- 말하기 어려움: 말이 불분명해지고, 점차 말하는 능력이 감소할 수 있습니다.
- 청력 손실: 일부 환자에서 청력 저하가 발생할 수 있습니다.
- 시각 장애: 시신경 위축으로 인해 시력 문제가 발생할 수 있습니다.
- 심장 질환: 심장 근육이 두꺼워지는 비후성 심근증이 발생할 수 있으며, 이는 생명을 위협할 수 있는 심각한 문제입니다.
- 당뇨병: 일부 환자에서 인슐린 생산에 문제가 발생할 수 있습니다.
진단
프레더릭 신경병증은 임상 증상, 가족력, 그리고 유전자 검사를 통해 진단됩니다. 유전자 검사는 FXN 유전자의 돌연변이 여부를 확인하여 확진할 수 있습니다.
치료
현재 프레더릭 신경병증을 완치할 수 있는 치료법은 없습니다. 치료는 증상을 관리하고 질병의 진행을 늦추는 데 중점을 둡니다. 일반적인 관리 방법은 다음과 같습니다:
- 물리 치료: 근육의 힘을 유지하고 관절의 유연성을 높이는 데 도움이 됩니다.
- 작업 치료: 일상 생활의 독립성을 유지하는 데 필요한 기술을 개발하는 데 도움이 됩니다.
- 언어 치료: 말하기 및 삼키기 능력을 개선하는 데 사용됩니다.
- 청력 보조 기기: 청력 손실이 있는 경우 사용할 수 있습니다.
- 심장 모니터링: 심장 질환의 조기 발견과 관리를 위해 정기적인 심장 검사가 필요합니다.
프레더릭 신경병증에 대한 연구는 계속되고 있으며, 새로운 치료법의 개발이 기대되고 있습니다.
2. 헌팅턴병 (Huntington's Disease)
헌팅턴병(Huntington's Disease, HD)은 중추신경계를 침범하는 유전적 질환으로, 특히 뇌의 특정 부분에 영향을 미치며 점진적인 운동, 인지 및 정신 장애를 초래합니다. 이 질환은 대개 중년기에 증상이 시작되며, 가족 세대를 거쳐 전달됩니다.
원인
헌팅턴병은 HTT 유전자의 돌연변이로 인해 발생합니다. 이 유전자는 헌팅틴이라는 단백질을 코딩하는데, 돌연변이로 인해 이 단백질의 일부분이 비정상적으로 길어집니다. 이 비정상적인 단백질이 뇌 세포에 축적되어 세포를 손상시키고 최종적으로는 세포의 사멸을 초래합니다. 헌팅턴병은 완전 우성 유전 패턴을 따르기 때문에, 돌연변이 유전자를 하나만 가지고 있어도 질병이 발현됩니다.
증상
헌팅턴병의 주요 증상은 크게 운동 장애, 인지 장애, 그리고 정신적 문제로 나눌 수 있습니다:
- 운동 장애: 초기에는 불안정하거나 불규칙한 자발적 움직임(코레아)이 특징입니다. 이는 점차 진행되어 일상적인 활동 수행이 어렵게 만듭니다.
- 인지 장애: 판단력과 계획 능력의 저하, 집중력 감소, 학습과 기억 문제 등이 발생합니다. 시간이 지남에 따라 이러한 증상은 점진적으로 악화됩니다.
- 정신적 문제: 우울증, 자극성 증가, 사회적 철수, 감정 조절 실패, 불안 및 환각이나 망상과 같은 정신증 증상이 나타날 수 있습니다.
진단
헌팅턴병의 진단은 주로 가족력, 임상 증상, 그리고 유전자 검사를 통해 이루어집니다. 유전자 검사는 HTT 유전자의 변이 부위인 CAG 반복 수를 측정하여 질병의 존재를 확진할 수 있습니다. CAG 반복 수가 비정상적으로 높은 경우 헌팅턴병 진단이 내려집니다.
치료
현재 헌팅턴병을 완치할 수 있는 치료법은 없습니다. 치료는 증상의 관리와 환자의 삶의 질 향상에 중점을 둡니다:
- 약물 치료: 운동 장애를 조절하는 약물(예: 도파민 길항제)과 우울증, 불안과 같은 정신 증상을 완화하는 약물이 사용됩니다.
- 물리 치료 및 작업 치료: 환자의 운동 능력과 일상 생활 활동을 최대한 유지하도록 도와줍니다.
- 언어 치료: 말하기와 삼키기 능력의 저하를 관리합니다.
- 심리적 지원: 환자와 가족을 대상으로 하는 상담이 중요하며, 질병의 진행 과정과 대처 방법에 대한 정보를 제공합니다.
헌팅턴병은 진행성 질환으로서, 환자와 가족에게 지속적인 의료 지원과 함께 감정적, 사회적 지원이 필요합니다.
3. 마르팡 증후군 (Marfan Syndrome)
마르팡 증후군(Marfan Syndrome)은 결합 조직의 유전적 장애로, 신체 전반의 여러 기관과 조직에 영향을 미칩니다. 특히 심장, 눈, 혈관, 뼈, 그리고 인대에 주로 영향을 주는 이 질환은 근골격계 이상, 시력 문제, 심장 질환 등을 초래할 수 있습니다.
원인
마르팡 증후군은 FBN1 유전자의 변이에 의해 발생합니다. 이 유전자는 피브릴린-1이라는 단백질을 생성하는데, 이 단백질은 결합 조직의 중요한 구성 요소입니다. 피브릴린-1의 결함은 결합 조직의 구조적 무결성에 영향을 미쳐 다양한 기관의 기능 장애를 초래합니다.
증상
마르팡 증후군의 증상은 다음과 같이 다양하게 나타날 수 있습니다:
- 근골격계 증상: 키가 비정상적으로 크고, 팔다리와 손가락이 긴 것이 특징입니다. 또한, 척추측만증(등이 휘는 증상), 가슴이 안으로 패이거나 밖으로 튀어나오는 등의 변형을 겪을 수 있습니다.
- 시각 증상: 렌즈 탈구(안구 내 렌즈가 정상 위치에서 벗어나는 증상)가 가장 흔하며, 근시가 매우 심한 경우가 많습니다.
- 심혈관 증상: 대동맥 확장이 가장 심각한 합병증 중 하나로, 이는 대동맥 파열로 이어질 수 있어 생명을 위협합니다. 또한, 심장 판막 이상이 나타날 수 있습니다.
- 폐 증상: 폐의 기능에 영향을 줄 수 있으며, 간혹 기흉(폐가 쪼그라드는 상태)이 발생할 수 있습니다.
진단
마르팡 증후군의 진단은 환자의 임상 증상, 가족력, 그리고 필요한 경우 유전자 검사를 통해 이루어집니다. FBN1 유전자의 돌연변이 확인은 진단을 확정하는 데 중요할 수 있으나, 증상과 신체 검사만으로도 진단이 가능한 경우가 많습니다.
치료
마르팡 증후군의 치료는 증상을 관리하고 가능한 합병증을 예방하는 데 중점을 둡니다:
- 약물 치료: 베타 차단제와 같은 약물을 통해 심장과 혈관의 부담을 줄일 수 있습니다.
- 수술적 치료: 대동맥 확장이 진행되거나 심각할 경우, 수술적 개입이 필요할 수 있습니다.
- 정기적인 모니터링: 정기적인 심장 및 눈 검사를 통해 조기에 문제를 발견하고 적절하게 대응합니다.
- 생활 습관 조절: 무거운 물건을 드는 것과 같은 신체적 스트레스를 피하고, 고강도 운동을 삼가는 것이 권장됩니다.
마르팡 증후군을 가진 개인과 가족에게는 전문적인 의료 지원과 교육이 필요합니다. 이 질환을 관리함에 있어 의료진과의 긴밀한 협력이 중요하며, 적절한 치료와 예방 조치를 통해 환자의 삶의 질을 유지하고, 장기적인 건강을 보호할 수 있습니다.
4. 외반모지증 (Ehlers-Danlos Syndrome)
유형
EDS에는 여러 하위 유형이 있으며, 가장 흔한 유형은 다음과 같습니다:
- 고전형 EDS: 피부의 과도한 신축성과 넓은 범위의 피부 상처가 특징입니다.
- 혈관형 EDS: 혈관과 내장 기관의 파열 위험이 높아 가장 위험할 수 있는 유형입니다.
- 고관절 유형 EDS: 주로 관절의 과도한 유연성과 빈번한 탈구를 겪습니다.
증상
EDS의 증상은 유형에 따라 다르지만, 일반적으로 다음과 같은 특징이 있습니다:
- 피부 증상: 피부가 매우 부드럽고 신축성이 뛰어나며, 쉽게 상처를 입고, 상처가 잘 아물지 않습니다.
- 관절 증상: 관절이 과도하게 유연하여 쉽게 탈구되고, 통증 및 만성 관절염을 유발할 수 있습니다.
- 혈관 증상: 특히 혈관형 EDS에서는 혈관이 취약해 쉽게 파열될 수 있으며, 이는 생명을 위협할 수 있습니다.
진단
EDS의 진단은 주로 임상 증상과 가족력에 기반하며, 유전 검사를 통해 특정 유형을 확인할 수 있습니다. 진단은 피부 또는 조직의 생검, 유전자 검사, 그리고 관련 증상에 대한 평가를 포함할 수 있습니다.
치료
현재 EDS를 치료할 수 있는 구체적인 방법은 없으며, 치료는 증상의 관리에 중점을 둡니다:
- 통증 관리: 비스테로이드성 항염증제(NSAIDs)와 같은 약물을 사용하여 관절과 근육의 통증을 관리할 수 있습니다.
- 물리치료: 관절의 안정성을 강화하고 탈구를 방지하기 위해 물리치료가 추천됩니다.
- 외과적 수술: 탈구가 잦거나 심각한 경우, 외과적 수술이 필요할 수 있습니다.
- 생활 습관 조절: 부상 방지를 위해 과도한 신체 활동을 피하고, 건강한 체중을 유지하는 것이 중요합니다.
예방 및 관리
EDS 환자와 가족은 질환에 대한 이해를 높이고, 적절한 생활 습관과 의료 관리를 통해 증상을 최소화하고 생활의 질을 향상시킬 수 있습니다. 정기적인 의료 검진과 전문가의 상담이 중요하며, 필요한 경우 유전 상담도 고려할 수 있습니다. EDS는 평생 관리가 필요한 만성 질환이기 때문에, 환자와 가족의 지속적인 교육과 지원이 필요합니다.
5. 리소좀 축적병 (Lysosomal Storage Disorders)

위 이미지는 리소좀 축적병(Lysosomal Storage Disorders)을 상세하게 묘사하고 있습니다. 여러 종류의 세포들—신경 세포, 간 세포, 근육 세포—이 표현되어 있으며, 각 세포 내에 리소좀에 축적된 물질들(지질, 뮤코다당류, 글리코프로틴)이 보입니다. 이 축적이 세포 손상과 기능 장애를 초래하는 모습이 강조되어 있습니다. 배경은 이 장애의 효소 결함과 유전적 측면을 교육적으로 설명하고 있습니다.
위 이미지는 리소좀 축적병(Lysosomal Storage Disorders)을 상세하게 묘사하고 있습니다. 여러 종류의 세포들—신경 세포, 간 세포, 근육 세포—이 표현되어 있으며, 각 세포 내에 리소좀에 축적된 물질들(지질, 뮤코다당류, 글리코프로틴)이 보입니다. 이 축적이 세포 손상과 기능 장애를 초래하는 모습이 강조되어 있습니다. 배경은 이 장애의 효소 결함과 유전적 측면을 교육적으로 설명하고 있습니다.
위 이미지는 리소좀 축적병(Lysosomal Storage Disorders)을 상세하게 묘사하고 있습니다. 여러 종류의 세포들—신경 세포, 간 세포, 근육 세포—이 표현되어 있으며, 각 세포 내에 리소좀에 축적된 물질들(지질, 뮤코다당류, 글리코프로틴)이 보입니다. 이 축적이 세포 손상과 기능 장애를 초래하는 모습이 강조되어 있습니다. 배경은 이 장애의 효소 결함과 유전적 측면을 교육적으로 설명하고 있습니다.
리소좀 축적병(Lysosomal Storage Disorders, LSDs)은 대사 장애의 한 그룹으로, 리소좀이라는 세포 내 소기관의 기능 이상 때문에 발생합니다. 리소좀은 세포의 "폐기 처리장" 역할을 하며, 단백질, 지방, 탄수화물 같은 큰 분자를 분해하고 재활용하는 데 중요합니다. 이러한 분해 작업에 필요한 효소가 결핍되거나 비활성화되면, 해당 물질들이 리소좀 내에 축적되어 세포 기능을 저해하고 다양한 임상 증상을 유발합니다.
원인
리소좀 축적병은 주로 유전적으로 발생하며, 특정 효소의 결핍이 질병의 직접적인 원인입니다. 이 효소들은 유전자 돌연변이로 인해 정상적으로 생성되지 않거나 비활성화됩니다. 대부분의 경우, 이 질환들은 자가 열성 유전 패턴을 따릅니다.
주요 유형
LSDs에는 50여 가지 이상의 다양한 질환이 포함되며, 가장 흔하거나 잘 알려진 몇 가지는 다음과 같습니다:
- 가우셔병 (Gaucher's Disease): 가장 흔한 형태로, 글루코세레브로시다아제라는 효소가 결핍되어 발생합니다.
- 파브리병 (Fabry Disease): 알파-갈락토시다제 A의 결핍으로 발생하며, 주로 남성에게 영향을 미칩니다.
- 헌터증후군 (Hunter Syndrome): 이다우로니다제의 결핍으로 인해 발생하며, 주로 남성에서 발견됩니다.
- 타이-삭스병 (Tay-Sachs Disease): 헥소사미나제 A의 결핍으로 중추신경계가 영향을 받습니다.
- 폼페병 (Pompe Disease): 알파-글루코시다제의 결핍으로 근육 및 심장 문제를 일으킵니다.
증상
리소좀 축적병의 증상은 매우 다양하고 각 질병마다 다릅니다. 일반적으로 이 질환들은 다음과 같은 증상을 포함할 수 있습니다:
- 발달 지연
- 운동 기능 저하
- 피부 및 눈의 변화
- 지적 장애
- 기관지 및 심장 문제
- 골격 이상
진단
진단은 임상 증상, 가족력, 그리고 생화학적 및 분자 유전학적 검사를 통해 이루어집니다. 효소 활성도 검사를 통해 특정 효소의 결핍을 확인하거나, 유전자 검사를 통해 관련 돌연변이를 확인할 수 있습니다.
치료
현재 리소좀 축적병에 대한 치료는 주로 증상 관리와 질환 진행의 지연에 초점을 맞춥니다. 일부 질환에 대해서는 효소 대체 요법(ERT)이 가능하며, 이는 결핍된 효소를 대체하는 정맥 주사 요법입니다. 유전자 치료, 줄기세포 이식 등의 치료법도 연구 중이거나 일부 질환에 적용되고 있습니다.
리소좀 축적병은 조기 진단과 적절한 관리가 중요한 질환군으로, 환자와 가족에게는 지속적인 의료 지원과 상담이 필요합니다.
6. 알젠트 증후군 (Alagille Syndrome)

알젠트 증후군(Alagille Syndrome)은 유전적 질환으로, 주로 간, 심장, 눈, 그리고 뼈에 영향을 미칩니다. 이 복합적인 장애는 특히 간의 담즙관이 비정상적으로 적어서 발생하는 담즙 울체와 관련이 있습니다. 이로 인해 간 기능 장애와 다양한 신체적 증상이 나타날 수 있습니다.
원인
알젠트 증후군은 대부분 JAG1 유전자의 돌연변이에 의해 발생합니다. 이 유전자는 세포 간 신호 전달에 중요한 역할을 하는 단백질을 생성하는데 필요하며, 이 단백질의 결핍이 여러 기관의 발달 장애를 초래합니다. 소수의 경우 NOTCH2 유전자의 변이도 이 질환과 연관될 수 있습니다.
증상
알젠트 증후군의 증상은 매우 다양하며 개인에 따라 차이가 큽니다. 일반적인 증상은 다음과 같습니다:
- 간 관련 증상: 담즙 울체로 인한 황달, 가려움증, 지방과 비타민의 흡수 장애
- 심장 질환: 심장의 혈관 이상, 특히 폐동맥 협착과 관련된 문제
- 눈의 변화: 포스터리어 조체 결손(Posterior embryotoxon)으로 눈의 조체 주변에 흰색 링이 형성되는 현상
- 골격 이상: 척추의 이상, 뼈의 변형 등
- 신장 이상과 혈관 이상: 신장 기능 장애, 신체의 다른 부위에 혈관 기형이 발생할 수 있음
진단
알젠트 증후군의 진단은 증상, 가족력, 그리고 유전자 검사를 통해 이루어집니다. 간 조직 검사를 통해 담즙관의 수가 비정상적으로 적은 것을 확인할 수 있으며, 심장 초음파나 안과 검사를 통해 다른 증상을 확인할 수 있습니다.
치료
알젠트 증후군의 치료는 증상에 따라 달라지며, 질환의 진행을 늦추고 삶의 질을 향상시키는 데 중점을 둡니다:
- 간 질환 관리: 증상에 따라 비타민 보충제와 지방 흡수를 돕는 약물을 사용합니다. 심한 경우 간 이식이 필요할 수 있습니다.
- 심장 질환 관리: 심장 협착이나 기타 문제에 대해 약물 치료 또는 수술적 개입이 필요할 수 있습니다.
- 가려움증 관리: 담즙산 바인더나 항히스타민제 등을 사용하여 증상을 완화할 수 있습니다.
- 정기적인 모니터링: 연령에 따라 정기적인 간 기능, 심장, 눈, 그리고 뼈 상태를 모니터링합니다.
알젠트 증후군은 생애 동안 지속적인 관리가 필요한 질환입니다.
7. 시스틴증 (Cystinosis)
시스틴증(Cystinosis)은 세포 내에서 특정 아미노산인 시스틴이 적절하게 제거되지 않아 생기는 희귀 대사 장애입니다. 이 질환은 세포의 리소좀에 시스틴이 축적되는 것이 특징으로, 시간이 지남에 따라 다양한 기관, 특히 신장에 심각한 손상을 줄 수 있습니다.
원인
시스틴증은 대부분 유전적 원인에 의해 발생합니다. 이 질환은 주로 CTNS 유전자의 돌연변이로 인해 발생하는데, 이 유전자는 시스틴을 세포에서 운반하는 역할을 하는 시스티노신 단백질을 코딩합니다. CTNS 유전자의 돌연변이로 인해 시스틴 운반체가 결핍되거나 비활성화되어 리소좀 내 시스틴이 축적되며, 이는 결국 세포 손상을 일으킵니다.
증상
시스틴증의 증상은 주로 세 가지 형태로 나타납니다:
- 유아형 시스틴증: 가장 흔하고 심각한 형태로, 생후 몇 달에서 몇 년 내에 증상이 나타납니다. 심한 탈수, 전해질 불균형, 신장 기능 장애, 저신장, 근육 약화, 갑상선 문제, 시력 문제(특히 결정질 침착) 등이 포함됩니다.
- 소아 및 청소년형 시스틴증: 더 늦은 나이에 증상이 나타나며, 유아형보다 경미할 수 있습니다.
- 성인형 시스틴증: 성인기에 발병하며 신장 문제가 주된 증상입니다.
진단
시스틴증의 진단은 임상 증상, 가족력, 그리고 세포 내 시스틴 수치 측정을 통해 이루어집니다. 특히 백혈구에서 시스틴 수치를 측정하는 것이 일반적입니다. 또한, 유전자 검사를 통해 CTNS 유전자의 변이를 확인할 수 있습니다.
치료
시스틴증의 치료는 주로 시스틴 수치를 감소시키고 기관 손상을 최소화하는 것을 목표로 합니다:
- 시스테아민: 이 약물은 시스틴 수치를 감소시키는 가장 효과적인 치료제로, 정기적으로 복용하면 리소좀 내 시스틴의 축적을 줄일 수 있습니다.
- 전해질 및 수분 보충: 신장 문제로 인한 전해질 불균형을 관리하기 위해 필요합니다.
- 지지 치료: 갑상선 기능 저하, 성장 장애, 근육 문제 등 다른 증상들에 대한 지지 치료가 필요할 수 있습니다.
- 정기적 모니터링: 주기적인 건강 검진을 통해 질병의 진행 상황을 모니터링하고, 필요한 치료 조정을 할 수 있습니다.
시스틴증은 조기 진단과 지속적인 치료가 중요한 질환으로, 적절한 치료와 관리를 통해 환자의 삶의 질과 예후를 크게 개선할 수 있습니다.
8. 가우셔병 (Gaucher's Disease)
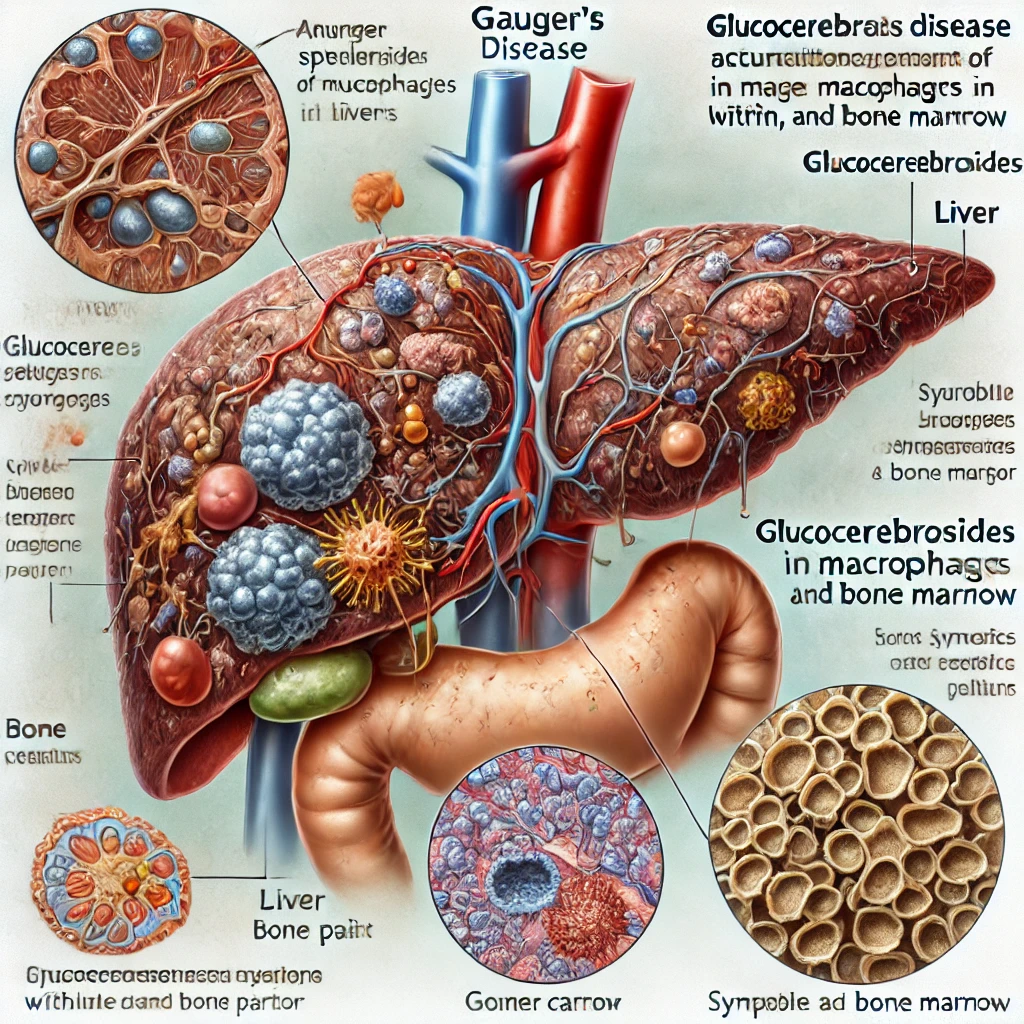
가우셔병(Gaucher's Disease)은 리소좀 축적병 중 가장 흔한 유형으로, 특정 지방 분해 효소의 결핍으로 인해 발생하는 유전적 장애입니다. 이 질환은 글루코세레브로시다아제라는 효소가 부족하거나 결핍되어, 글루코세레브로사이드라는 지방이 세포 내 리소좀에 축적되는 것이 특징입니다. 이 지방 축적은 주로 간, 비장, 뼈, 그리고 때로는 골수에 영향을 미칩니다.
증상
가우셔병의 증상은 세 가지 주요 유형으로 나뉘며 각 유형별로 증상이 다르게 나타납니다:
- 1형 가우셔병: 가장 흔한 형태로, 비장과 간이 커지는 것이 특징입니다. 이로 인해 복부 팽창, 피로, 빈혈, 출혈 경향 증가 및 뼈 문제(통증 및 골절)가 발생할 수 있습니다. 유럽계 유대인에게서 가장 흔히 발견됩니다.
- 2형 가우셔병: 생후 몇 개월 내에 증상이 나타나며, 매우 심각하고 급속하게 진행됩니다. 신경계 증상을 포함하여 일반적으로 생후 2년 이내에 생명을 위협할 수 있습니다.
- 3형 가우셔병: 신경계 증상이 점진적으로 나타나며, 눈의 움직임 장애(가로 안진증)와 같은 신경학적 문제가 특징입니다. 이 형태는 또한 신체의 다른 부분에도 영향을 미칩니다.
진단
가우셔병의 진단은 임상 증상, 가족력, 그리고 글루코세레브로시다아제 효소 활성도 검사를 통해 이루어집니다. 유전자 검사를 통해 GBA 유전자의 돌연변이를 확인할 수 있으며, 이는 진단을 확정짓는 데 도움이 됩니다.
치료
가우셔병의 치료는 주로 질병의 유형과 진행 정도에 따라 달라집니다:
- 효소 대체 요법(ERT): 결핍된 글루코세레브로시다아제를 대체하기 위해 정기적으로 효소를 주사하는 방법입니다. 이 치료는 특히 1형 가우셔병 환자에게 효과적이며, 비장과 간의 크기를 줄이고, 혈액 수치를 개선하며, 뼈 문제를 완화할 수 있습니다.
- 경구용 약물: 일부 새로운 치료제는 효소 대체 요법의 대안으로 경구 투여가 가능하며, 이러한 약물은 글루코세레브로시다아제의 기능을 보조하거나 글루코세레브로사이드의 생성을 감소시킵니다.
- 증상 관리: 뼈 통증 및 기타 증상 관리를 위해 추가적인 약물 치료가 필요할 수 있습니다. 또한, 정기적인 의료 모니터링이 중요합니다.
가우셔병은 유전적 질환으로 평생 관리가 필요할 수 있으며, 적절한 치료와 지속적인 의료 지원을 통해 많은 환자들이 삶의 질을 향상시키고 있습니다.
9. 파브리병 (Fabry Disease)
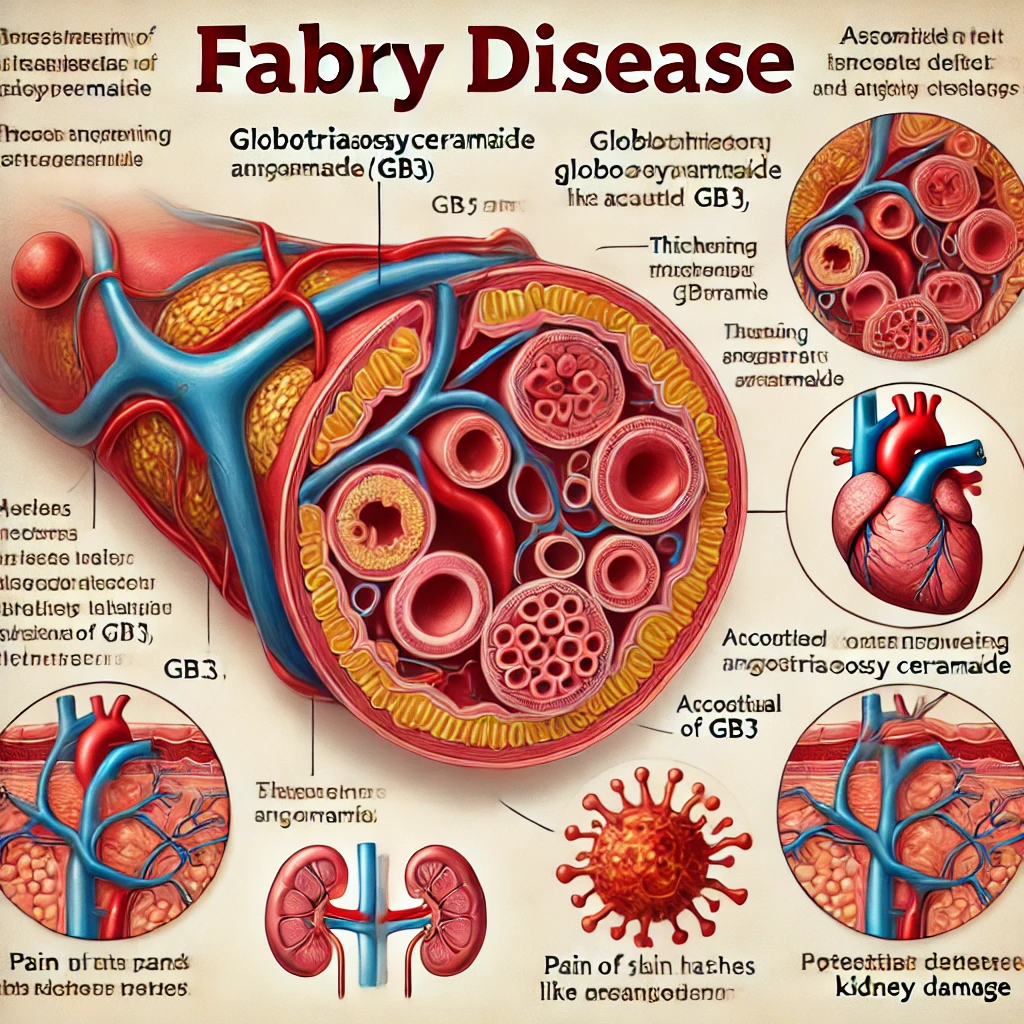
파브리병(Fabry Disease)은 리소좀 축적 질환의 한 형태로, 성염색체 열성 유전 방식으로 전달되는 대사 장애입니다. 이 질환은 알파-갈락토시다제 A(α-galactosidase A) 효소의 결핍이 원인으로, 이 효소의 부족은 지방 분자인 글로보트리아오실세라마이드(GL-3 또는 Gb3)가 세포 내 리소좀에 축적되게 합니다. 이 축적은 다양한 조직과 장기에 영향을 미치며, 특히 신장, 심장, 피부 및 신경계에 문제를 일으킬 수 있습니다.
증상
파브리병의 증상은 개인마다 다양하며, 남성에서 더 심각하고 일찍 나타나는 경향이 있습니다. 주요 증상은 다음과 같습니다:
- 통증: 손과 발에 발생하는 극심한 통증(발작성 통증)이 특징적입니다.
- 피부 이상: 소형 붉은 발진(앵지오케라토마)이 주로 배, 엉덩이, 대퇴부에 나타납니다.
- 신장 기능 장애: 만성 신장 질환으로 진행되며, 심각한 경우 신장 기능 손실로 이어질 수 있습니다.
- 심혈관 문제: 심장 질환, 불규칙한 심장 박동, 심장 마비가 발생할 수 있습니다.
- 소화 기능 장애: 복통, 설사, 불규칙한 배변 등 소화기 증상이 나타날 수 있습니다.
- 시력 및 청력 문제: 안구의 혈관 변화로 시력 감소가 발생할 수 있고, 청력 손실도 가능합니다.
진단
파브리병의 진단은 효소 활성 검사로 알파-갈락토시다제 A의 활성 수준을 측정함으로써 확인할 수 있습니다. 여성의 경우 이 효소의 활성이 정상 범위 내에서 변동할 수 있기 때문에, 유전자 검사가 더 확실한 진단 방법으로 권장됩니다.
치료
파브리병의 치료는 증상의 관리와 질병의 진행을 늦추는 데 초점을 맞춥니다. 주요 치료 방법은 다음과 같습니다:
- 효소 대체 요법(ERT): 결핍된 알파-갈락토시다제 A 효소를 대체하는 정맥 주사를 통해 신체의 효소 수준을 보충합니다.
- 경구용 약물 치료: Migalastat와 같은 경구용 약물이 일부 환자에게 사용될 수 있으며, 이 약물은 효소의 안정성을 증가시키고 기능을 개선합니다.
- 증상에 대한 지지적 치료: 통증 관리, 심혈관 건강 유지, 신장 기능 모니터링 및 지원이 포함됩니다.
파브리병은 평생 관리가 필요한 만성 질환이며, 정기적인 의료 모니터링과 포괄적인 관리 계획이 환자의 삶의 질을 유지하고 개선하는 데 중요합니다.
10. 엘러스-단로스 증후군 (Ehlers-Danlos Syndrome)
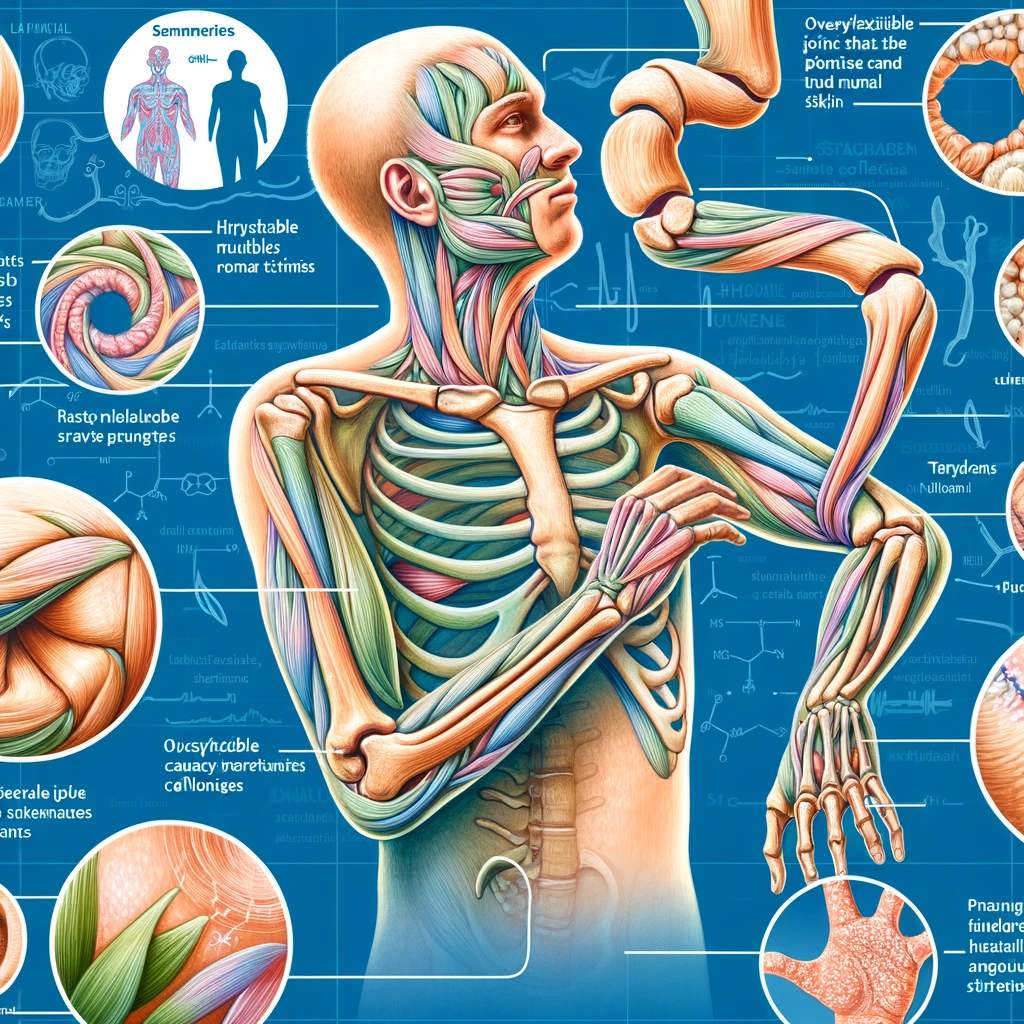
엘러스-단로스 증후군(Ehlers-Danlos Syndrome, EDS)은 다양한 유형이 있는 결합 조직 장애로, 콜라겐의 이상으로 인해 피부, 관절, 혈관 등에 영향을 미치는 질환입니다. 콜라겐은 신체의 결합 조직을 강화하고 지지하는 데 필요한 주요 단백질로, 이 단백질의 결함은 많은 구조적 문제를 일으킬 수 있습니다.
증상
EDS의 증상은 해당 유형에 따라 다양하지만, 일반적인 증상은 다음과 같습니다:
- 피부: 피부가 비정상적으로 신축성이 좋고, 부드럽고 쉽게 멍이 듭니다. 상처가 잘 아물지 않으며, 때로는 흉터가 지거나 탄력이 떨어집니다.
- 관절: 관절이 과도하게 유연하여 쉽게 탈구되거나 과신전됩니다. 이로 인해 통증과 만성 관절염이 발생할 수 있습니다.
- 혈관: 일부 EDS 유형(특히 혈관형)은 혈관이 취약하여 쉽게 파열될 수 있으며, 이는 생명을 위협할 수 있습니다.
유형
EDS는 여러 하위 유형으로 분류됩니다. 가장 흔한 유형은 다음과 같습니다:
- 고전형 EDS: 피부와 관절 증상이 두드러지며, 피부가 매우 신축성이 있고 쉽게 상처를 입습니다.
- 혈관형 EDS: 가장 심각한 유형으로, 주로 혈관과 내장 기관의 파열 위험이 있습니다.
- 고관절형 EDS: 관절의 과신전과 관련된 증상이 주를 이루며, 일부 환자에서는 조기에 관절염이 발생할 수 있습니다.
진단
EDS의 진단은 주로 임상 증상, 가족력, 그리고 필요한 경우 유전자 검사를 통해 이루어집니다. 진단은 종종 피부 생검, 관절 및 피부의 신축성 평가, 그리고 혈관 이미징 검사를 포함할 수 있습니다. 유전자 검사는 특정 유형의 EDS를 확인하는 데 도움이 될 수 있으며, 많은 경우에 가족 내 다른 사람들의 진단에도 중요합니다.
치료
EDS의 치료는 주로 증상의 관리에 중점을 둡니다:
- 통증 관리: 비스테로이드성 항염증제(NSAIDs)를 포함한 약물을 사용하여 통증을 관리합니다.
- 물리치료: 관절의 안정성을 유지하고 부상을 예방하기 위해 물리치료가 추천됩니다.
- 외과적 개입: 필요한 경우, 탈구된 관절의 수술적 교정이나 혈관 합병증의 수술적 치료가 이루어질 수 있습니다.
- 생활 습관 조정: 과도한 신체 활동을 피하고, 적절한 영양 섭취와 건강한 생활 습관을 유지하는 것이 중요합니다.
EDS 환자는 다양한 의료 전문가의 지속적인 관리가 필요하며, 적절한 치료와 지원을 통해 삶의 질을 향상시키고 가능한 합병증을 예방할 수 있습니다.
11. 폼페병 (Pompe Disease)

폼페병(Pompe Disease)은 리소좀 축적 질환 중 하나로, 알파-글루코시다제(lysosomal alpha-glucosidase, GAA)라는 특정 효소의 결핍 또는 부족으로 인해 발생하는 유전적 장애입니다. 이 효소는 글리코겐을 포도당으로 분해하는 역할을 합니다. 효소의 부족은 글리코겐이 근육세포의 리소좀에 축적되어 근육 및 신경 세포에 손상을 입히는 결과를 초래합니다.
유형
폼페병은 발병 시기에 따라 크게 두 가지 형태로 분류됩니다:
- 유아형 폼페병(Infantile-onset Pompe Disease): 생후 몇 개월 내에 증상이 나타나며, 심한 근육 약화, 심장 문제, 그리고 조기 사망으로 이어질 수 있습니다.
- 성인형 폼페병(Late-onset Pompe Disease): 어린이기, 청소년기 또는 성인기에 시작되며, 주로 근육 약화와 호흡 곤란을 특징으로 합니다.
증상
폼페병의 증상은 유형에 따라 다르며 다음과 같습니다:
- 유아형:
- 심한 근육 약화
- 심장 비대(심장근증)
- 먹이를 먹기 어려움
- 발달 지연
- 호흡곤란
- 성인형:
- 점진적인 근육 약화, 특히 다리와 몸통에 영향을 미침
- 호흡곤란, 특히 누워 있을 때 악화
- 피로
- 운동 능력 저하
진단
폼페병의 진단은 증상, 가족력, 효소 활성도 검사, 그리고 유전자 검사를 통해 이루어집니다. 혈액 샘플에서 GAA 효소의 활성도를 측정하여 효소 결핍을 확인하며, 유전자 검사를 통해 진단을 확정할 수 있습니다.
치료
폼페병의 치료는 다음과 같습니다:
- 효소 대체 요법(Enzyme Replacement Therapy, ERT): 결핍된 GAA 효소를 대체하기 위한 정기적인 정맥 주사입니다. 이 치료는 특히 유아형에서 생존율과 생활의 질을 개선하는 데 효과적입니다.
- 대증 치료: 호흡 보조기기 사용, 물리 치료, 그리고 다른 증상에 대한 지지적 관리가 포함됩니다.
- 규칙적인 모니터링: 심장과 호흡 기능의 정기적인 평가가 중요하며, 증상의 변화에 따라 치료 계획을 조정해야 합니다.
폼페병은 생명을 위협할 수 있는 질환으로, 조기 진단과 적극적인 치료 접근이 중요합니다.
12. 타르 증후군 (TAR Syndrome)

타르 증후군(Thrombocytopenia-Absent Radius Syndrome, TAR Syndrome)은 선천적인 희귀 질환으로, 주로 혈소판 감소증(Thrombocytopenia)과 두 팔의 요골(방사골, Radius) 부재를 특징으로 합니다. 이 질환은 추가적인 골격 이상 및 다른 신체적 특징들을 동반할 수 있습니다.
증상
타르 증후군의 주요 증상은 다음과 같습니다:
- 혈소판 감소증: 생후 초기에 나타나며, 출혈 경향이 높아집니다. 이는 멍, 출혈, 그리고 때때로 심각한 출혈 문제를 일으킬 수 있습니다.
- 팔의 요골 부재: 양쪽 팔의 요골이 없는 상태로 태어나며, 이로 인해 팔이 비정상적으로 구부러지고 기능에 제한을 받을 수 있습니다. 그러나 상완골(위팔뼈)은 일반적으로 존재합니다.
- 기타 골격 이상: 무릎, 엉덩이, 척추에도 이상이 발생할 수 있으며, 발달 상의 지연이나 다른 형태의 골격 문제가 동반될 수 있습니다.
- 신장 이상 및 심장 문제: 일부 환자에서는 신장 기형 또는 심장 결함이 보고되기도 합니다.
원인
타르 증후군은 유전적 요인에 의해 발생합니다. 최근의 연구에 따르면 RBM8A 유전자의 돌연변이가 이 질환과 관련이 있는 것으로 밝혀졌습니다. 이 유전자의 변이는 일반적으로 열성 유전 패턴을 따르지만, 추가적인 환경적 또는 유전적 요인이 질환의 발현에 영향을 미칠 수 있습니다.
진단
타르 증후군의 진단은 임상 증상과 가족력을 기반으로 이루어집니다. 진단을 확정하기 위해 유전학적 검사, X-레이, 그리고 혈액 검사를 통해 혈소판 수를 확인할 수 있습니다. 때로는 태아기 초음파를 통해 진단이 이루어지기도 합니다.
치료
타르 증후군의 치료는 증상에 따라 다르게 접근합니다:
- 혈소판 수 관리: 혈소판 수가 매우 낮은 경우 혈소판 수혈을 통해 출혈 위험을 관리합니다.
- 정형외과적 중재: 팔과 다른 관절의 기형 교정을 위한 수술이 필요할 수 있습니다.
- 지지적 치료: 물리 치료와 작업 치료를 통해 환자의 기능적 능력을 향상시키고 일상생활 활동을 돕습니다.
- 정기적인 모니터링: 출혈 경향, 심장 및 신장 기능을 정기적으로 검사하여 관련 건강 문제를 조기에 발견하고 관리합니다.
타르 증후군은 평생 지속되는 관리가 필요한 질환으로, 다학제 팀이 관여하는 포괄적인 의료 접근 방식이 필요합니다.
13. 윌슨병 (Wilson's Disease)
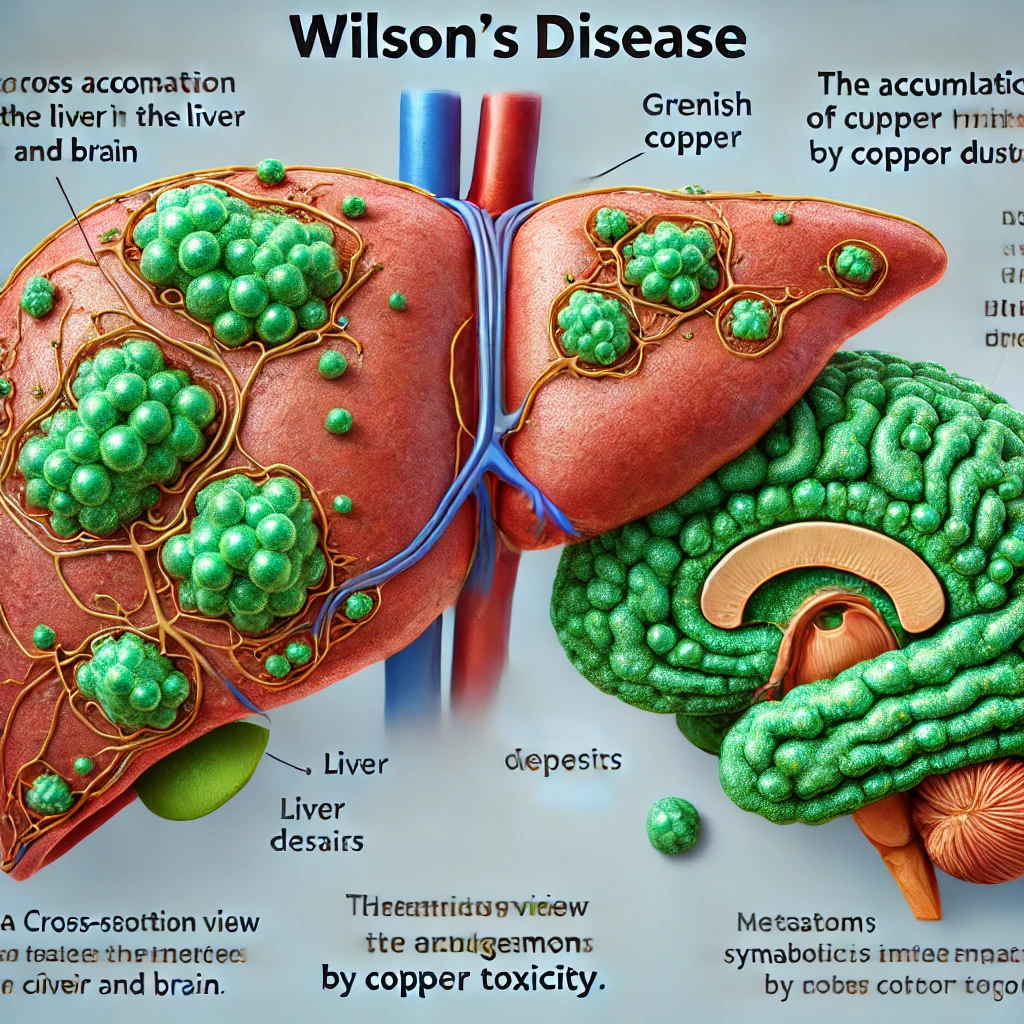
윌슨병(Wilson's Disease)은 유전적으로 발생하는 희귀 대사 장애로, 체내에서 구리의 대사와 배출이 제대로 이루어지지 않아 체내에 구리가 과도하게 축적되는 것이 특징입니다. 이 과도한 구리는 간, 뇌, 신장 및 기타 장기에 손상을 입힐 수 있으며, 적절히 치료하지 않을 경우 심각한 건강 문제나 생명을 위협할 수 있습니다.
원인
윌슨병은 ATP7B 유전자의 돌연변이에 의해 발생합니다. 이 유전자는 간에서 구리를 대사하고 체외로 배출하는 데 중요한 역할을 하는 단백질을 생성하는 데 필요합니다. 유전자의 돌연변이로 인해 이 단백질이 제대로 기능하지 못하면, 구리가 간에 축적되기 시작하며 결국 다른 장기로 이동하여 손상을 일으킬 수 있습니다.
증상
윌슨병의 증상은 다양하며, 주로 간 손상과 신경학적 장애로 나타납니다:
- 간 질환: 피로, 식욕 부진, 간 기능 장애, 황달, 복부 팽창 등이 발생할 수 있습니다.
- 신경학적 증상: 손 떨림, 걸음걸이 이상, 언어 장애, 기분 변화, 우울증 등이 나타날 수 있으며, 심한 경우 근육 강직이나 경련이 발생할 수 있습니다.
- 기타 증상: 신장 결석, 관절 통증, 빈혈 등이 발생할 수 있으며, 눈의 각막 가장자리에 동반되는 케이저-플라이셔 환(Kayser-Fleischer rings)라고 불리는 동그란 갈색 또는 녹색의 환 형태로 나타날 수 있습니다.
진단
윌슨병의 진단은 혈액 검사, 간 기능 검사, 소변에서의 구리 수치 측정, 각막의 케이저-플라이셔 환 검사, 간 조직 검사 및 유전자 검사를 통해 이루어질 수 있습니다. 이 질환의 조기 진단과 치료는 중요합니다.
치료
윌슨병의 치료는 구리 축적을 감소시키고 증상을 관리하는 것을 목표로 합니다:
- 구리 킬레이트제: D-페니실아민(D-penicillamine) 또는 트리엔틴(Trientine)과 같은 약물을 사용하여 체내의 구리를 결합하고 배출시키는 방법입니다.
- 아연 보충제: 아연은 구리의 흡수를 방해하여 체내 구리 수치를 낮출 수 있습니다.
- 식이 요법: 구리가 풍부한 식품(예: 간, 견과류, 초콜릿, 버섯, 조개류 등)의 섭취를 제한합니다.
- 간 이식: 심각한 간 손상의 경우, 간 이식이 필요할 수 있습니다.
윌슨병은 평생 관리가 필요한 질환입니다.
14. 버거병 (Buerger's Disease)

버거병(Buerger's Disease), 또는 혈전성 혈관염증성병(Thromboangiitis Obliterans)은 주로 손과 발의 작은 및 중간 크기 혈관(동맥과 정맥)에 영향을 미치는 질환입니다. 이 병은 혈관의 염증과 혈전(피떡) 형성을 특징으로 하며, 이로 인해 혈류가 감소되거나 차단되어 심각한 통증과 감염, 궁극적으로는 조직의 괴사를 유발할 수 있습니다.
원인
버거병의 정확한 원인은 아직 명확히 밝혀지지 않았습니다. 하지만, 흡연은 버거병 발병과 밀접한 관련이 있으며, 거의 모든 환자가 흡연자 또는 전 흡연자입니다. 흡연은 혈관 염증을 유발하고 혈전 형성을 촉진하는 것으로 생각됩니다. 또한 유전적 요인, 면역 체계의 이상, 감염 등이 다른 가능한 원인으로 제시되고 있습니다.
증상
버거병의 증상은 다음과 같습니다:
- 손발의 통증 및 저림: 특히 활동 중이나 야간에 증상이 심해질 수 있습니다.
- 손과 발의 차가움: 혈류 감소로 인해 손발이 차가워질 수 있습니다.
- 상처 회복 지연: 손발에 생긴 상처가 잘 아물지 않습니다.
- 색 변화: 손발의 피부 색이 창백해지거나 푸르스름해지고, 때로는 붉어질 수 있습니다.
- 궤양 형성 및 조직 괴사: 질환의 진행으로 손가락이나 발가락에 궤양이 발생하거나 괴사할 수 있습니다.
진단
버거병의 진단은 주로 증상과 환자의 흡연력에 기반하며, 혈관 조영술, 초음파 검사, MRI, 혈액 검사 등을 통해 다른 질환과 구별됩니다. 혈관 조영술은 혈관의 염증과 혈전이 있는 위치를 명확히 보여줄 수 있습니다.
치료
버거병의 치료는 주로 증상의 관리와 질환 진행의 예방에 중점을 둡니다:
- 금연: 버거병을 가진 모든 환자에게 필수적이며, 증상의 개선과 질환의 진행을 막을 수 있는 가장 중요한 조치입니다.
- 혈관 확장제: 혈관을 확장시켜 혈류를 개선하는 약물을 사용할 수 있습니다.
- 통증 관리: 비스테로이드성 항염증제(NSAIDs)와 같은 통증 조절 약물이 사용될 수 있습니다.
- 조직 손상 관리: 궤양 치료와 감염 방지를 위한 적절한 상처 관리가 필요합니다.
- 수술적 치료: 혈관 우회술 또는 괴사된 조직의 제거가 필요한 경우 수술적 개입이 이루어질 수 있습니다.
버거병은 질환의 예후를 크게 개선할 수 있는 금연과 같은 생활 습관의 변화가 중요한 질환입니다.
15. 스티븐스-존슨 증후군 (Stevens-Johnson Syndrome)

스티븐스-존슨 증후군(Stevens-Johnson Syndrome, SJS)은 피부와 점막에 심각한 반응을 일으키는 드물고 위험한 장애입니다. SJS는 피부의 광범위한 손상과 함께 물집이 생기고 피부가 벗겨지는 것이 특징입니다. 이 상태는 종종 특정 약물의 사용 또는 감염에 대한 반응으로 발생하며, 즉시 치료하지 않으면 생명을 위협할 수 있습니다.
원인
스티븐스-존슨 증후군의 정확한 원인은 항상 명확하지 않지만, 주요 원인으로는 다음과 같은 것들이 있습니다:
- 약물: 항생제(설폰아미드), 항경련제, NSAIDs(비스테로이드성 항염증제), 알로푸리놀 등의 약물이 SJS를 유발할 수 있습니다.
- 감염: 헤르페스 바이러스, 인플루엔자, HIV 등과 같은 감염이 SJS의 트리거가 될 수 있습니다.
- 유전적 요인: 일부 유전적 소인이 SJS의 위험을 증가시킬 수 있습니다.
증상
SJS의 증상은 다음과 같이 발전합니다:
- 초기 증상: 발열, 불쾌감, 기침, 두통, 목 아픔 등 감기와 비슷한 증상이 나타납니다.
- 피부 증상: 통증이 있는 빨간색 또는 자주색 발진이 몸 전체에 급속하게 퍼지며, 몇 시간 내에 물집이 생기고 피부가 벗겨질 수 있습니다.
- 점막 손상: 입, 눈, 비뇨기계, 성기 등의 점막에도 물집과 궤양이 발생합니다.
진단
SJS의 진단은 주로 임상 증상과 환자의 의약품 사용력, 최근 감염력 등의 병력에 근거합니다. 피부 생검을 통해 다른 피부 질환과 구별할 수 있으며, 진단을 확정하기 위해 필요할 수 있습니다.
치료
SJS의 치료는 주로 지지적 치료와 증상 관리에 중점을 둡니다:
- 병원 입원: SJS는 종종 중환자실에서 치료를 필요로 합니다.
- 약물 중단: SJS를 유발한 것으로 의심되는 약물은 즉시 중단합니다.
- 통증 관리 및 수액 보충: 통증을 관리하고 충분한 수분을 섭취하도록 합니다.
- 상처 치료: 물집과 궤양이 있는 피부와 점막을 치료하고 감염을 예방합니다.
- 영양 지원: 영양 상태를 유지하고 회복을 돕기 위해 필요할 수 있습니다.
스티븐스-존슨 증후군은 의료적 긴급 상황으로, 즉각적인 치료가 필요합니다.
16. 아크로데르마티티스 엔테로파티카 (Acrodermatitis Enteropathica)
아크로데르마티스 엔테로파티카(Acrodermatitis Enteropathica)는 아연 흡수 장애로 인해 발생하는 희귀한 유전적 대사 장애입니다. 이 질환은 체내에서 아연의 흡수와 대사가 제대로 이루어지지 않아 아연 결핍을 초래하며, 피부 발진, 설사, 그리고 면역계 문제를 일으킬 수 있습니다.
원인
아크로데르마티스 엔테로파티카는 대부분 유전적 원인에 의해 발생하며, SLC39A4 유전자의 돌연변이로 인해 발생합니다. 이 유전자는 아연을 세포 내로 운반하는 중요한 단백질을 코딩하는데, 돌연변이로 인해 이 단백질의 기능이 손상되어 장에서 아연의 흡수가 제대로 이루어지지 않습니다. 이 질환은 자가 열성 유전 질환으로, 부모로부터 두 복사본의 돌연변이 유전자를 물려받을 경우 발병합니다.
증상
아크로데르마티스 엔테로파티카의 주요 증상은 다음과 같습니다:
- 피부 발진: 손, 발, 얼굴 및 기저귀 지역에서 발생하는 피부 발진이 일반적입니다. 이 발진은 종종 염증을 동반하고, 습진이나 궤양으로 발전할 수 있습니다.
- 소화 장애: 심한 설사와 복통이 발생할 수 있으며, 이는 영양소 흡수 장애로 이어질 수 있습니다.
- 성장 지연: 어린이에서는 성장 지연이 발생할 수 있습니다.
- 면역계 약화: 빈번한 감염과 일반적인 면역계 약화 증상이 나타날 수 있습니다.
진단
진단은 증상, 가족력, 그리고 혈중 아연 수치 검사를 포함할 수 있습니다. 증상과 저혈중 아연 수치가 진단에 결정적인 역할을 합니다. 유전자 검사를 통해 SLC39A4 유전자의 돌연변이를 확인할 수 있습니다.
치료
아크로데르마티스 엔테로파티카의 치료는 주로 아연 보충을 통해 이루어집니다:
- 아연 보충제: 아연 결핍을 교정하기 위해 아연 보충제를 경구로 섭취합니다. 이 치료는 증상의 개선을 빠르게 가져올 수 있으며, 평생 지속될 수 있습니다.
- 지지적 치료: 피부 관리와 영양 지원이 필요할 수 있으며, 감염 예방을 위한 추가적인 조치가 필요할 수 있습니다.
이 질환은 적절한 치료로 대부분의 증상이 잘 관리될 수 있으며, 아연 보충을 통해 환자는 정상적인 삶을 영위할 수 있습니다.
17. 알렉산더병 (Alexander Disease)
알렉산더병(Alexander Disease)은 매우 드문 신경 변성 질환으로, 주로 중추신경계의 교질세포에 영향을 미칩니다. 교질세포는 뇌와 척수에 존재하는 지지 세포로, 신경계의 정상적인 기능을 돕는 중요한 역할을 합니다. 알렉산더병은 주로 유전적 요인에 의해 발생하며, GFAP(Glial Fibrillary Acidic Protein) 유전자의 돌연변이가 주요 원인으로 알려져 있습니다. 이 단백질의 이상은 신경계 내에서 비정상적인 단백질 축적을 유발하며, 이로 인해 심각한 신경 합병증이 발생합니다.
증상
알렉산더병의 증상은 환자의 나이와 질환의 형태에 따라 다르게 나타납니다. 이 질환은 주로 세 가지 형태로 분류됩니다:
- 유아형(Infantile): 가장 흔한 형태로, 보통 생후 2년 이내에 증상이 시작됩니다. 증상으로는 머리 크기 증가(대두증), 발달 지연, 발작, 강직 및 먹이 섭취 문제 등이 있습니다.
- 소아-청소년형(Juvenile): 증상은 보통 어린이나 청소년기에 시작되며, 발달 지연, 조정 운동 장애, 언어 및 학습 문제 등이 포함됩니다.
- 성인형(Adult): 성인에서 시작되며, 보행 장애, 언어 문제, 인지 장애 등이 점진적으로 발생합니다.
진단
알렉산더병의 진단은 임상 증상, 영상 진단 결과(MRI 등), 그리고 필요한 경우 유전자 검사를 통해 이루어집니다. MRI 검사는 뇌의 비정상적인 변화를 보여줄 수 있으며, 특히 교질세포의 이상이 두드러지게 나타납니다. 유전자 검사는 GFAP 유전자의 돌연변이를 확인하여 진단을 확정할 수 있습니다.
치료
현재 알렉산더병을 치료할 수 있는 특정 치료법은 없습니다. 치료는 주로 증상의 관리와 삶의 질 향상에 중점을 둡니다:
- 지지적 치료: 영양 관리, 물리치료, 작업치료 등이 포함됩니다.
- 약물 치료: 발작 관리를 위한 항경련제와 기타 증상을 완화하기 위한 약물이 사용될 수 있습니다.
- 정기적인 모니터링: 질병의 진행 상황을 모니터링하고 적절한 치료 조정을 위해 정기적인 의료 검진이 필요합니다.
알렉산더병은 가족과 환자 모두에게 큰 도전이 될 수 있는 질환입니다. 따라서 환자와 가족이 질환에 대해 잘 이해하고, 필요한 지원을 받을 수 있도록 의료진, 상담사, 지역 사회 자원과의 긴밀한 협력이 중요합니다.
18. 아미로이드증 (Amyloidosis)
아미로이드증(Amyloidosis)은 비정상적인 단백질, 아미로이드가 조직이나 장기에 축적되어 발생하는 질환군을 말합니다. 이 비정상적인 단백질 축적은 장기의 구조와 기능을 방해하여 다양한 건강 문제를 일으킬 수 있습니다. 아미로이드증은 다양한 형태가 있으며, 각 형태는 축적되는 아미로이드 단백질의 종류와 영향을 받는 장기에 따라 달라집니다.
아미로이드증의 주요 유형
- 원발성 아미로이드증 (AL amyloidosis): 이전에 '경량 체인 질환'으로 알려진 이 유형은 골수에서 비정상적인 면역 세포가 과도하게 증식하여 경량 단백질 체인이 생성되고, 이 체인이 조직에 축적되는 것이 특징입니다. 주로 심장, 신장, 간, 신경계 등에 영향을 미칩니다.
- 연관성 아미로이드증 (AA amyloidosis): 만성 염증성 질환(예: 류마티스 관절염, 염증성 장 질환 등)과 관련이 있으며, 염증 과정에서 생성되는 단백질이 장기에 축적됩니다.
- 유전성 아미로이드증: 유전적 변이로 인해 특정 단백질이 비정상적으로 접히고 축적되는 것이 특징입니다. 예를 들어, 트랜스티레틴 아미로이드증은 주로 심장이나 신경계에 영향을 미칩니다.
- 노인성 아미로이드증: 나이가 들면서 심장에 특정 아미로이드 단백질이 축적되는 현상으로, 심장 기능 저하를 일으킬 수 있습니다.
- 투석 관련 아미로이드증: 장기간 투석을 받는 환자에서 볼 수 있는 형태로, 베타2-마이크로글로불린이 축적되어 관절과 힘줄에 문제를 일으킵니다.
증상
아미로이드증의 증상은 축적 위치와 장기의 종류에 따라 다양합니다:
- 심장: 심장 부정맥, 심부전, 흉통
- 신장: 단백뇨, 신부전, 부종
- 신경계: 감각 이상, 통증, 근력 약화
- 소화계: 구역, 체중 감소, 변비 또는 설사
- 기타: 피로, 체중 감소, 빈혈
진단
아미로이드증의 진단은 생검을 통해 이루어집니다. 영향을 받은 조직에서 아미로이드 단백질의 존재를 확정하는 것이 중요합니다. 또한 혈액 검사, 소변 검사, 이미징 검사(예: 심장 초음파, MRI)를 통해 진단을 보조할 수 있습니다.
치료
아미로이드증의 치료는 주로 증상의 관리와 기저 질환의 치료에 초점을 맞춥니다. 원발성 아미로이드증의 경우 화학 요법이나 줄기세포 이식과 같은 치료가 사용될 수 있습니다. AA 아미로이드증은 염증을 조절하는 치료가 중요하며, 유전성 아미로이드증은 특정 유전자 치료나 약물을 사용할 수 있습니다. 모든 형태의 아미로이드증에 대해 지속적인 모니터링과 적절한 의료 관리가 필요합니다.
19. 프레그릴리 X 증후군 (Fragile X Syndrome)

프레그릴리 X 증후군(Fragile X Syndrome, FXS)은 X 염색체에 있는 FMR1 유전자의 돌연변이 때문에 발생하는 유전적 장애입니다. 이 증후군은 지적 장애, 행동 문제, 그리고 특정한 신체적 특징들을 특징으로 하며, 발달 장애 중 가장 흔한 유전적 원인 중 하나로 알려져 있습니다. 특히 남성에서 더 자주 발견되며, 여성보다 증상이 더 심각한 경향이 있습니다.
원인
프레그릴리 X 증후군은 FMR1 유전자의 CGG 염기서열 반복이 비정상적으로 많아질 때 발생합니다. 정상적인 경우, 이 반복은 5~44회 정도이지만, 프레그릴리 X 증후군 환자에서는 200회 이상 반복될 수 있습니다. 이러한 과도한 반복은 유전자의 기능을 방해하여 FMRP(프레그릴리 X 정신 지체 단백질)의 생산을 감소시킵니다. FMRP는 뇌에서 중요한 역할을 하는 단백질로, 특히 학습과 기억에 관여합니다.
증상
프레그릴리 X 증후군의 증상은 매우 다양할 수 있습니다:
- 지적 장애: 대부분의 남성 환자에서 경미에서 중증의 지적 장애가 나타납니다. 여성 환자의 경우 증상이 더 경미하거나 존재하지 않을 수 있습니다.
- 행동 문제: 주의력 결핍, 과잉행동, 불안, 사회적 상호작용에 어려움, 감각 과민 등이 포함됩니다.
- 신체적 특징: 긴 얼굴, 큰 귀, 늘어진 눈꺼풀, 유연한 관절, 남성에서 큰 고환이 관찰될 수 있습니다.
- 언어 장애: 언어 발달 지연이 흔하며, 말이 빠르고 반복적일 수 있습니다.
진단
프레그릴리 X 증후군의 진단은 임상 증상을 바탕으로 하며, 유전자 검사를 통해 확정됩니다. 유전자 검사는 FMR1 유전자 내의 CGG 반복 횟수를 측정하여 진단을 내립니다.
치료
현재 프레그릴리 X 증후군을 완치할 수 있는 치료법은 없습니다. 치료는 증상을 완화하고 환자의 삶의 질을 향상시키는 데 중점을 둡니다:
- 교육적 지원: 맞춤형 교육 프로그램과 특수 교육이 필요할 수 있습니다.
- 행동 치료: 행동 문제를 관리하기 위한 전략이 적용됩니다.
- 언어 치료: 의사소통 기술을 개선하기 위해 필요합니다.
- 약물 치료: 불안, 주의력 결핍 및 기타 관련 증상을 관리하기 위해 약물이 사용될 수 있습니다.
프레그릴리 X 증후군에 대한 이해와 관리는 지속적인 의료 및 교육적 지원을 통해 이루어져야 하며, 환자와 가족 모두에게 지속적인 지원이 중요합니다.
이 질환들은 각기 다른 신체 시스템에 영향을 미치며, 이에 따라 다양한 증상과 합병증을 나타냅니다. 일부는 생명을 위협할 수 있고, 일부는 관리 가능할 수 있습니다. 대부분의 희귀질환은 유전적 요인에 의해 발생하며, 정확한 진단과 적절한 치료가 필수적입니다.
희귀질환에 대한 연구는 계속해서 진행 중이며, 새로운 치료법과 기술의 발전이 이러한 질환을 앓고 있는 환자들에게 새로운 희망을 제공하고 있습니다. 그럼에도 불구하고, 많은 희귀질환은 여전히 도전적인 상황에 처해 있으며, 이에 대한 지속적인 연구와 지원이 필요합니다.
Store : https://smartstore.naver.com/searchroad - 스마트스토어
Youtube : https://www.youtube.com/channel/UCdEfOUEgB98CIhitcVc42Pw - 힐링유튜브
https://www.youtube.com/channel/UClw00TgE71lrCbdMSWDI1UQ- 힐링유튜브
출처: https://happyadvisor.tistory.com/75 [잡다정보공유:티스토리]
헬스장 부럽지 않은 홈짐 IM.GYM : 써치로드 (naver.com)
헬스장 부럽지 않은 홈짐 IM.GYM : 써치로드
[써치로드] 좋은 제품, 믿을 수 있는 제품을 많이 소개할 수 있는 스토어가 되었으면 합니다
smartstore.naver.com


"이 포스팅은 쿠팡 파트너스 활동의 일환으로, 이에 따른 일정액의 수수료를 제공받습니다."
'건강정보' 카테고리의 다른 글
| 당신의 속쓰림, 역류성 식도염일 수 있습니다: 증상부터 치료까지 (0) | 2024.08.03 |
|---|---|
| 집에서 할 수 있는 짧은 운동 루틴 5가지! (2) | 2024.07.30 |
| 카페인의 두 얼굴: 우리 몸에 미치는 효과와 부작용 (0) | 2024.05.27 |
| 급성 알코올 중독의 이해와 대처 (4) | 2024.05.27 |
| 뎅기열: 모기가 전하는 열대병 (0) | 2024.05.14 |